



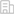

華東師范大學生命科學學院吳宇軒、劉明耀、李大力團隊、中南大學湘雅醫院付斌團隊和邦耀生物合作報告了世界首例CRISPR基因編輯治療β0/β0型重度地貧兒童患者的臨床結果。截至日前,兩例受試者脫離輸血依賴均已超過24個月。
β-地中海貧血是一種遺傳性溶血性疾病,在世界范圍內普遍流行,是最常見的單基因疾病之一1。因功能性β-珠蛋白嚴重缺乏,相當一部分患者需要定期輸血才能存活,從而導致輸血依賴性地中海貧血(TDT)。鑒于血液資源有限和鐵螯合劑成本高昂,國內TDT患者僅少部分能維持規范輸血和規范去鐵治療,生存狀態堪憂,TDT患者的生存率明顯低于發達國家2,3。
由于兒童和青少年患者對造血干細胞移植(HSCT)的治療相關毒性具有更好的耐受性,符合條件的患者應盡早接受 HSCT(2-7 歲)4。但對于發展中國家和欠發達地區兒童,探索基因編輯治療兒童 TDT 至關重要并且尤為緊迫。
基因編輯破壞+58 BCL11A紅系增強子的GATA1結合位點可誘導γ-珠蛋白表達,這是緩解HBB基因突變引起的β-血紅蛋白病的一種頗有前景的治療策略5-7。在本研究中,研究人員報告了一項正在進行的 1/2 期試驗 (NCT04211480) 的初步結果,該試驗評估了基因編輯治療對輸血依賴性β-地中海貧血 (TDT) 兒科患者的安全性和有效性。
該試驗將基因編輯的自體造血干祖細胞(HSPCs)移植到兩名兒童患者體內,其中一名患兒基因型為β0/β0,被列為最嚴重的TDT類型;另外一例接受治療的患兒也屬于TDT類型。
截至文章投稿時,兩名患兒的胎兒血紅蛋白分別從基線時的2.55g/L、1.75g/L分別上升至最近一次訪視的149g/L和139g/L,最近一次訪視時的總血紅蛋白含量分別為152g/L和140g/L。
截至文章投稿時,兩例患者在治療后都實現了脫離輸血依賴超過16個月,脫離輸血依賴的定義為為輸血的情況下總血紅蛋白達到或超過90g/L。移植后第9個月時的骨髓細胞中的編輯率分別為85.46%和89.48%。
患者在移植后分別于 52 天和40 天出院,最近一次訪視時臨床狀況良好。清髓預處理相關毒性較輕,不良反應包括 1-2 級粘膜炎、1 級皮疹和 2 級鼻出血。細胞移植之后沒有發生嚴重的感染,鐵過載情況也有明顯改善。
兩名患者的紅細胞數量和總體血紅蛋白水平在第 45 天左右開始穩定增加,在第 75 天左右達到健康水平。
截至日前,兩例受試者脫離輸血依賴均已超過24個月。
移植后紅細胞和血紅蛋白變化趨勢,F細胞占比以及鐵代謝數據
對編輯后重建的PBMCs插入缺失模式的探索性分析顯示,在近兩年的隨訪期內,沒有觀察到異常的克隆擴增現象。同時,研發人員對經基因編輯后重建的PBMCs進行了單細胞RNA測序,全面分析由未編輯或編輯的HSPC重組的血液譜系的轉錄組,表明BCL11A紅系特異增強子編輯不會導致非紅細胞的顯著轉錄變化,不會影響B細胞以及DC細胞的發育和功能相關基因的表達,這些結果均顯示該療法沒有明顯的副作用。
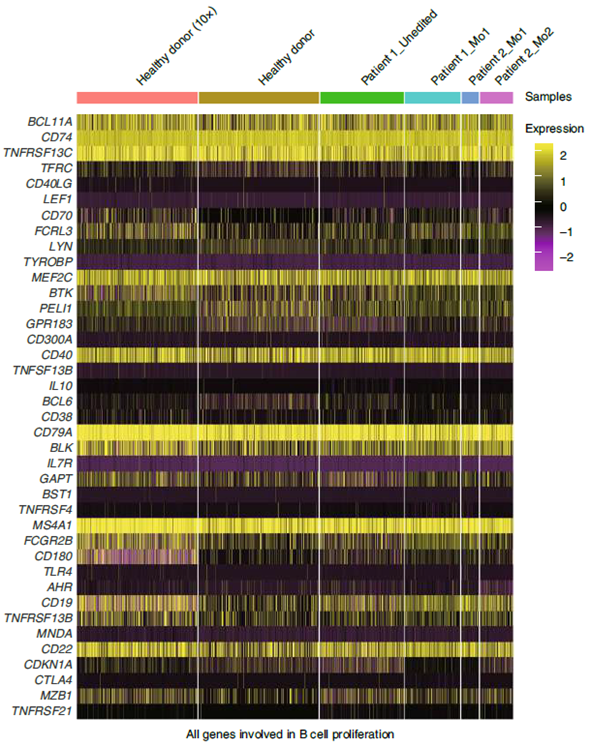
健康供體和經基因編輯后病人的B細胞簇中B細胞增殖相關基因的表達
總之,該研究提供了可以實現 CRISPR/Cas9 編輯的自體HSPC的移植和長期植入的原理證明,并且證實了胎兒血紅蛋白水平的持續升高足以改善輸血依賴性β-地中海貧血,即使對于β-珠蛋白鏈生成完全受抑制的β0/β0 基因型也是如此。
該成果在 Nature Medicine 雜志上發表,題為 CRISPR–Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia 。華東師大生命科學學院吳宇軒教授、劉明耀教授和李大力教授為論文共同通訊作者;中南大學湘雅醫院付斌教授、華東師大生命科學學院吳宇軒課題組廖嬌陽博士和陳雙紅博士是本文的共同第一作者。中南大學湘雅醫院為論文第一完成單位,華東師大為第二完成單位,研究過程中還得到了S. Siwko博士和哈佛醫學院、波士頓兒童醫院的D. E. Bauer博士的大力幫助。同時感謝參與本項研究的受試者及其家人的支持和配合。